
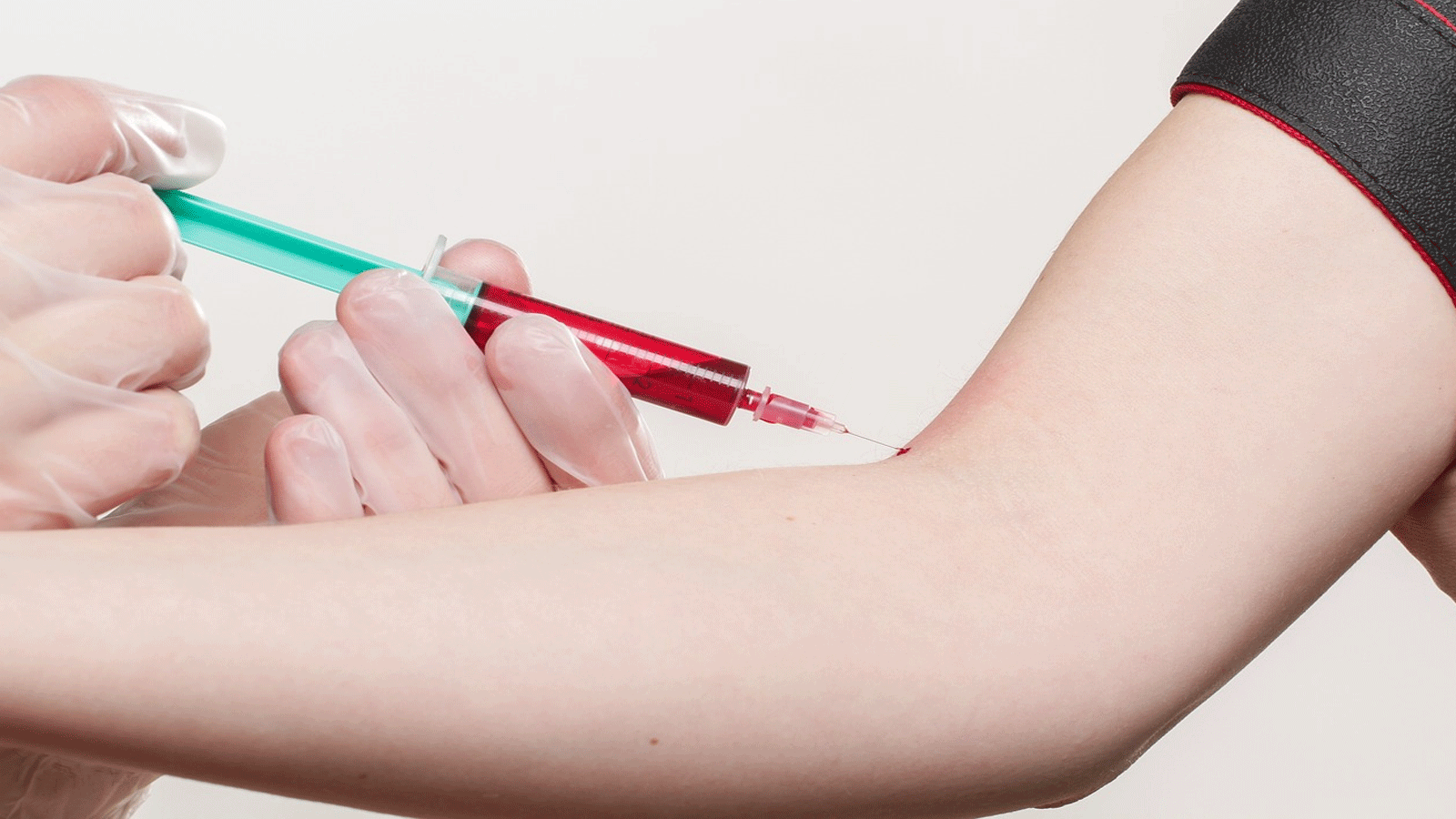
أعلنت اللجنة المستقلة المساعدة لإدارة الغذاء والدواء الأميركية، والمكونة من مجموعة خبراء، ان العلاج الجيني الثوري المقترح لمرض فقر الدم المنجلي "exa-cel" آمن بدرجة كافية.
وتوقعت أن توافق هيئة الغذاء والدواء على استخدامه بحلول الثامن من ديسمبر المقبل.
ويمكن لهذا الدواء أن يعالج الأشخاص من مرض فقر الدم المنجلي المؤلم والمميت الذي يعاني منه حوالى 100 ألف شخص أميركي، وينتشر أيضا في دول أخرى في الشرق الأوسط وأفريقيا.
ويعمل هذا العلاج على تعديل الجينات المعروفة باسم "CRISPR"، بحسب شبكة "سي إن إن" الأميركية.
وكانت إدارة الغذاء والدواء الأميركية قد اعترفت بفعالية العلاج الجيني الذي تم تطويره بشكلٍ مشترك من قبل شركة "فيرتكس" للأدوية، التي تتخذ من بوسطن مقرا لها، وشركة "CRISPR Therapeutics" السويسرية.
وأجرت "فيرتكس" تجارب سريرية على هذا العلاج الثوري، شملت 44 مريضًا وتمت متابعة 30 منهم فقط لمدة 16 شهرًا على الأقل.
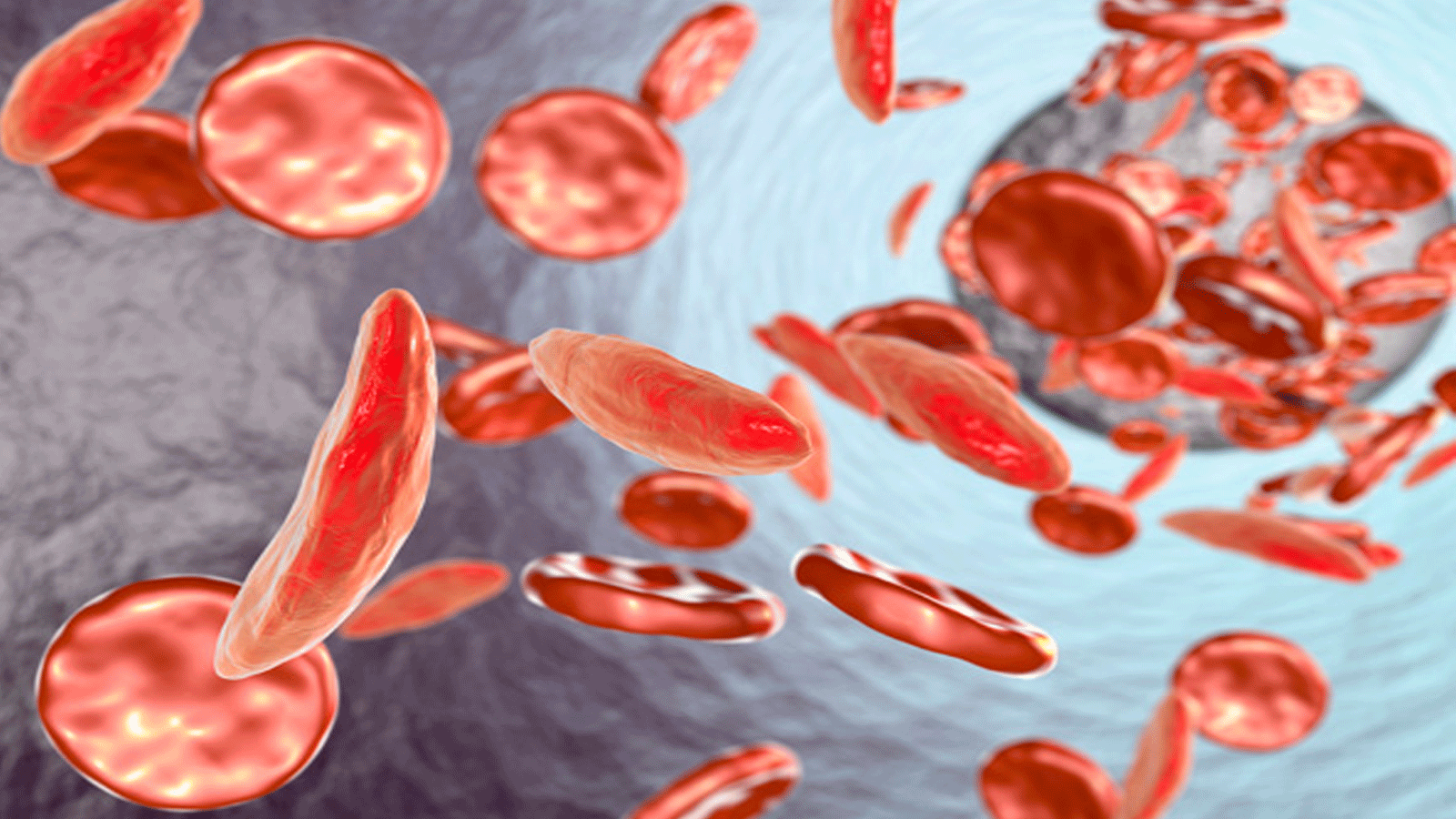
صورة تشرح كيفية تشكل كريات الدم لدى مرضى فقر الدم المنجلي نشرتها شركة pfizer للأدوية
فقر الدم المنجلي
وينتمي فقر الدم المنجلي إلى مجموعة من الاضطرابات تُعرف باسم مرض الخلايا المنجلية، وتؤثر هذه الحالة على شكل خلايا كريات الدم الحمراء، التي تحمل الأوكسجين إلى كل أجزاء الجسم.
فبينما تكون خلايا الدم الحمراء الطبيعة مستديرة ومرنة، مما يسهل حركتها في الأوعية الدموية، تأخذ الكريات شكل الهلال أو المنجل لدى المصابين بهذا المرض الوراثي وتصبِح صلبة ولزجة.
وهو ما يسبب آلامًا للمرضى بسبب بطء تدفق الدم داخل الأوعية الدموية.
يشار إلى أن إدارة الغذاء والدواء الأميركية ستقر استخدام علاج محتمل ثاني لمرض الخلية المنجلية بحلول 20 ديسمبر.
وهو علاج جيني آخر ابتكرته شركة "بلوبيرد بيو" في ولاية ماساشوستس.
وقالت إدارة الغذاء والدواء الأميركية إن علاج الخلايا فقر الدم المنجلي هو "حاجة طبية لم تتم تلبيتها".
ومن المتوقع أن تكون كلفة هذه العلاجات الجديدة عالية جدا، وربما ستكلف ملايين الدولارات لكل مريض، لكن الرعاية مدى الحياة للمصابين بهذا المرض تكلف نظام الرعاية الصحية في الولايات المتحدة ما يقدر بنحو 3 مليارات دولار سنويا، وفقا لـ"نيويورك تايمز". ما يعني ان اكتشاف هذا العلاج سيجد حلاً لأزمة بقيت بدون علاج على مدى عقود.








التعليقات